Defining a Genome-wide TR Catalog
Most existing tools for genotyping tandem repeats (TRs) from short-read or long-read sequencing data require the user to provide the exact start/end coordinates and motifs of all TR loci to genotype. Due to this, a key step in any genome-wide TR analysis is to define the loci to include (aka. the TR catalog). This choice directly affects sensitivity since any loci not in the catalog will be missing from the downstream analysis. Also, it determines compute costs since runtimes of most current tools are proportional to the number of loci being genotyped. There is no consensus in the field on how to choose a catalog, with recent papers and presentations all using very different sets of loci as their starting points.
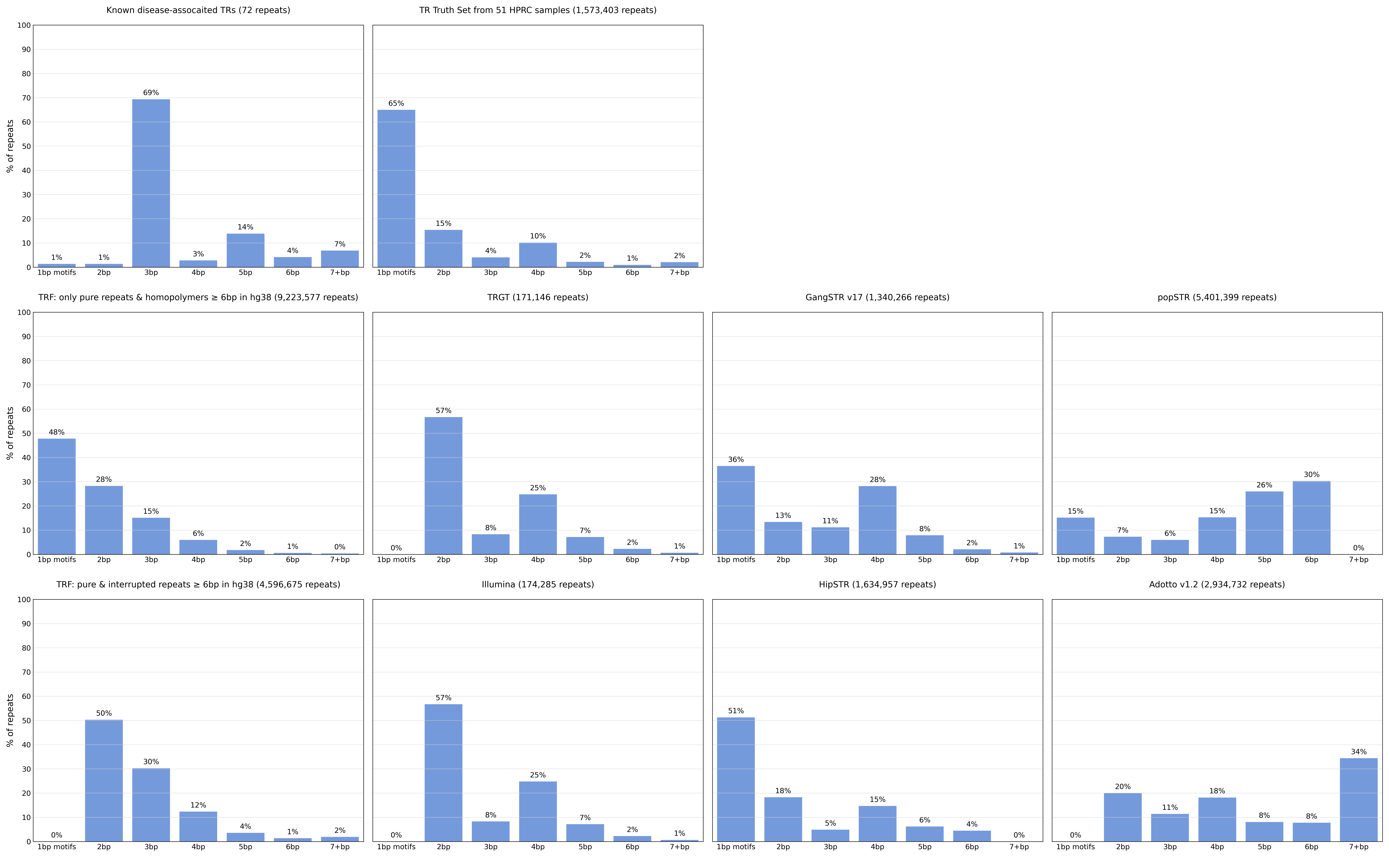
Catalogs of TR loci can differ in multiple ways, including:
- total # of loci
- whether they include homopolymers
- their overall distribution of motif sizes
- inclusion of chrX or chrY
- whether they include interrupted repeats, and if yes, how often their locus definitions overlap each other
** The counts represent repeat intervals (aka. loci) in each of the catalogs after filtering out a small numbers of erroneous records from several of the catalogs (such as homopolymers of 'N' rather than A/C/G/T).
** Some catalogs - such as the known disease-associated loci catalog - define adjacent repeats. For example, FXN is defined as (A)*(GAA)* rathern than just (GAA)*. I counted any adjacent repeats as separate loci in the stats below, which is why the catalog of known disease-assocaited TRs is counted as having a small number of homopolymer loci.
| catalog | total loci | chrX | chrY | homo- polymers? |
motif sizes |
locus sizes |
# of repeats | pure repeats
|
trimmed
|
overlapping loci
|
|---|---|---|---|---|---|---|---|---|---|---|
| Known disease-associated loci | 72 | 8 (11.3%) | 0 (0.0%) | 1-27bp | 12-150bp | 1-50x | 58.3% | 100.0% | 0 (0.0%) | |
| TRF catalog: only pure repeats + homopolymers spanning ≥ 6bp in hg38 |
9,223,577 | 489,574 (5.3%) | 0 (0.0%) | 1-50bp | 6-1597bp | 3-300x | 100.0% | 78.0% | 0 (0.0%) | |
| GangSTR v17 | 1,340,266 | 65,719 (4.9%) | 8,979 (0.7%) | 1-20bp | 10-612bp | 3-248x | 100.0% | 100.0% | 16 (0.0%) | |
| TRF catalog: pure & interrupted repeats spanning ≥ 6bp in hg38 |
4,596,675 | 244,906 (5.3%) | 0 (0.0%) | 2-50bp | 6-6800bp | 3-1091x | 85.1% | 100.0% | 84,822 (1.8%) | |
| HipSTR | 1,634,957 | 79,233 (4.8%) | 12,638 (0.8%) | 1-6bp | 11-102545bp | 3-20509x | 46.6% | 100.0% | 0 (0.0%) | |
| TRGT | 171,146 | 8,558 (5.0%) | 0 (0.0%) | 2-24bp | 4-532bp | 2-133x | 95.5% | 100.0% | 0 (0.0%) | |
| Illumina | 174,285 | 8,561 (4.9%) | 0 (0.0%) | 2-24bp | 4-532bp | 2-133x | 95.5% | 100.0% | 0 (0.0%) | |
| TR truth set from 51 HPRC samples |
1,573,403 | 70,885 (4.5%) | 4,806 (0.3%) | 1-50bp | 1-1591bp | 1-301x | 94.7% | 100.0% | 131,515 (8.4%) | |
| popSTR | 5,401,399 | 0 (0.0%) | 0 (0.0%) | 1-6bp | 11-137bp | 2-128x | 78.0% | 42.0% | 11,202 (0.2%) | |
| Adotto v1.2 | 2,934,732 | 109,740 (3.7%) | 16,585 (0.6%) | 2-495bp | 4-49816bp | 1-8836x | 41.6% | 32.0% | 26,198 (0.9%) | |
| Platinum TRs v1.0 | 7,722,729 | 328,981 (5.2%) | 47,192 (0.7%) | 1-822bp | 4-9545bp | 1-2659x | 62.2% | 36.1% | 0 (0.0%) |
Motif size distribution in each catalog:
Generating a combined catalog:
I think that currently the best way to create a comprehensive genome-wide TR catalog is to merge:- several of the large catalogs generated by running TRF on the reference with different parameters (ie. only pure repeats, and separately, repeats with mismatches but not indels). These catalogs will cover a mix of loci that are a) highly polymorphic b) rarely polymorphic (eg. differ from the reference genome in < 1/10,000 individuals), and c) not polymorphic at all (ie. the same in all humans).
- all available empirical catalogs - including the Illumina catalog and the TR truth set from the table above - since these were generated using orthogonal methods that don't require some minimum number of repeats in the reference genome. These catalogs will include polymorphic loci that have only 1 or 2 repeats in hg38, and so are not captured by TRF-based catalogs.
Combined catalogs:
I generated two combined catalogs - a smaller one with 4.7M loci and a larger one with 9.8M loci. The only difference between them is whether they include TRF-generated catalogs of loci that span at least 6bp in the reference, or at least 9bp. Both combined catalogs are publicly available for download:| catalog | total loci | chrX | chrY | homo- polymers? |
motif sizes |
locus sizes |
# of repeats | pure repeats
|
trimmed
|
overlapping loci
|
|---|---|---|---|---|---|---|---|---|---|---|
| Known disease-associated loci | 72 | 8 (11.3%) | 0 (0.0%) | 1-27bp | 12-150bp | 1-50x | 58.3% | 100.0% | 0 (0.0%) |
Technical details:
The str-analysis repo contains scripts for merging, annotating and filtering TR catalogs.The commands below use these scripts to generate a combined TR catalog:
## download the publicly-available catalogs from the google buckets and repos from [weisburd 2023]
known_disease_associated_loci=https://raw.githubusercontent.com/broadinstitute/str-analysis/main/str_analysis/variant_catalogs/variant_catalog_without_offtargets.GRCh38.json
trf_pure_6bp_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/repeat_specs_GRCh38_without_mismatches.including_homopolymers.sorted.at_least_6bp.bed.gz
trf_pure_9bp_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/repeat_specs_GRCh38_without_mismatches.including_homopolymers.sorted.at_least_9bp.bed.gz
gangstr_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/hg38_ver17.adjusted.bed.gz
trgt_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/trgt_repeat_catalog.hg38.reformatted_to_motif_only.bed.gz
illumina_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/illumina_variant_catalog.sorted.bed.gz
truth_set_catalog=https://storage.cloud.google.com/str-truth-set-v2/results_with_homopolymers/results_from_combined/combined.51_samples.positive_loci.json
hipstr_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/hg38.hipstr_reference.adjusted.bed.gz
trf_6bp_catalog_with_mismatches=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/repeat_specs_GRCh38_allowing_mismatches.sorted.trimmed.at_least_6bp.bed.gz
trf_9bp_catalog_with_mismatches=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/repeat_specs_GRCh38_allowing_mismatches.sorted.trimmed.at_least_9bp.bed.gz
popstr_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/popstr_catalog_v2.bed.gz
adotto_catalog=https://storage.cloud.google.com/str-truth-set/hg38/ref/other/adotto_tr_catalog_v1.2.bed.gz
ref_fasta=/local/path/hg38.fa
output_prefix=combined_catalog.trf_at_least_9bp
## annotate each catalog and discard loci with N in the motif
for path in ${known_disease_associated_loci} \
${trf_pure_9bp_catalog} \
${gangstr_catalog} \
${trf_9bp_catalog_with_mismatches} \
${hipstr_catalog} \
${trgt_catalog} \
${illumina_catalog} \
${truth_set_catalog} \
${popstr_catalog} \
${adotto_catalog};
do
# download each catalog
wget $path
# annotate loci and discard edge cases like motif=N
python3 -u -m str_analysis.annotate_and_filter_str_catalog \
-r ${ref_fasta} \
--output-stats \
--output-tsv \
--verbose \
--discard-loci-with-non-acgt-bases \
${path}
done
## merge all catalogs into a single file (excluding the popSTR and Adotto catalogs because they are unusual and/or suboptimal in some ways)
python3 -u -m str_analysis.combine_str_catalogs --verbose \
--merge-adjacent-loci-with-same-motif \
--output-prefix ${output_prefix} \
--add-source-field \
$(basename ${known_disease_associated_loci} | sed 's/.json/.annotated_and_filtered.json.gz/') \
$(basename ${trf_pure_9bp_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${gangstr_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${trf_9bp_catalog_with_mismatches} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${hipstr_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${trgt_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${illumina_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${truth_set_catalog} | sed 's/.json/.annotated_and_filtered.json.gz/')
## add adjacent loci to the catalog
python3 -u -m str_analysis.add_adjacent_loci_to_expansion_hunter_catalog \
--ref-fasta ${ref_fasta} \
--max-distance-between-adjacent-repeats 6 \
--max-adjacent-repeats 2 \
--source-of-adjacent-loci ~/code/str-truth-set/ref/other/repeat_specs_GRCh38_without_mismatches.including_homopolymers.sorted.at_least_6bp.bed.gz \
${output_prefix}.json.gz
## annotate the combined catalog
python3 -u -m str_analysis.annotate_and_filter_str_catalog \
-r ${ref_fasta} \
--output-stats \
--output-tsv \
--verbose \
--discard-loci-with-non-acgt-bases \
${output_prefix}.with_adjacent_loci.json.gz
## compute catalog stats
python3 -u -m str_analysis.compute_catalog_stats --verbose \
$(basename ${known_disease_associated_loci} | sed 's/.json/.annotated_and_filtered.json.gz/') \
$(basename ${trf_pure_6bp_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${gangstr_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${popstr_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${trf_6bp_catalog_with_mismatches} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${hipstr_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${trgt_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${illumina_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/') \
$(basename ${truth_set_catalog} | sed 's/.json/.annotated_and_filtered.json.gz/') \
$(basename ${adotto_catalog} | sed 's/.bed.gz/.annotated_and_filtered.json.gz/')
Running these steps yields a combined catalog with the following properties:
Stats for combined_catalog.trf_at_least_9bp.with_adjacent_loci.annotated_and_filtered.json.gz:
4,918,783 total loci
1,139,440 out of 4,918,783 ( 23.2%) loci have adjacent repeats
6,196,806 total repeat intervals
4,522,377 out of 6,196,806 ( 73.0%) repeat interval size is an integer multiple of the motif size (aka. trimmed)
1,792,664 out of 6,196,806 ( 28.9%) repeat intervals are homopolymers
1,534,528 out of 6,196,806 ( 24.8%) repeat intervals overlap each other by at least two motif lengths
Examples of overlapping repeats: chr19:22862456-22862510, chr16:87038648-87038702, chr6:131131459-131131467, chr3:75804442-75804495, chrX:53113326-53113335, chrX:141112874-141112888, chr14:36902984-36902999, chr12:6740055-6740065, chr16:7165935-7165979, chr15:91057135-91057191
Ranges:
Motif size range: 1-50bp
Locus size range: 1-102545bp
Num repeats range: 1-20509x repeats
Maximum locus size = 102545bp @ chrY:11490351-11592896 (AATGG)
Minimum fraction pure bases = 0.00 @ chrY:26670808-26670828 (AATGG)
Minimum fraction pure repeats = 0.00 @ chrX:155370990-155371126 (TATATAACTATAAATAAGTATATATTTATATATT)
Minimum overall mappability = 0.00 @ chrY:24489213-24489225 (GCCT)
chrX: 322,966 out of 6,196,806 ( 5.2%) repeat intervals
chrY: 16,670 out of 6,196,806 ( 0.3%) repeat intervals
chrM: 15 out of 6,196,806 ( 0.0%) repeat intervals
alt contigs: 0 out of 6,196,806 ( 0.0%) repeat intervals
Motif size distribution:
1bp: 1,792,664 out of 6,196,806 ( 28.9%) repeat intervals
2bp: 1,479,000 out of 6,196,806 ( 23.9%) repeat intervals
3bp: 1,532,929 out of 6,196,806 ( 24.7%) repeat intervals
4bp: 839,986 out of 6,196,806 ( 13.6%) repeat intervals
5bp: 256,601 out of 6,196,806 ( 4.1%) repeat intervals
6bp: 126,362 out of 6,196,806 ( 2.0%) repeat intervals
7-24bp: 120,964 out of 6,196,806 ( 2.0%) repeat intervals
25+bp: 48,300 out of 6,196,806 ( 0.8%) repeat intervals
Fraction pure bases distribution:
0.0: 8,522 out of 6,196,806 ( 0.1%) repeat intervals
0.1: 8,595 out of 6,196,806 ( 0.1%) repeat intervals
0.2: 16,730 out of 6,196,806 ( 0.3%) repeat intervals
0.3: 16,591 out of 6,196,806 ( 0.3%) repeat intervals
0.4: 15,682 out of 6,196,806 ( 0.3%) repeat intervals
0.5: 36,774 out of 6,196,806 ( 0.6%) repeat intervals
0.6: 58,015 out of 6,196,806 ( 0.9%) repeat intervals
0.7: 82,843 out of 6,196,806 ( 1.3%) repeat intervals
0.8: 141,233 out of 6,196,806 ( 2.3%) repeat intervals
0.9: 71,488 out of 6,196,806 ( 1.2%) repeat intervals
1.0: 5,740,333 out of 6,196,806 ( 92.6%) repeat intervals
Mappability distribution:
0.0: 30,591 out of 4,918,783 ( 0.6%) loci
0.1: 21,219 out of 4,918,783 ( 0.4%) loci
0.2: 27,035 out of 4,918,783 ( 0.5%) loci
0.3: 29,659 out of 4,918,783 ( 0.6%) loci
0.4: 38,958 out of 4,918,783 ( 0.8%) loci
0.5: 68,023 out of 4,918,783 ( 1.4%) loci
0.6: 51,683 out of 4,918,783 ( 1.1%) loci
0.7: 70,885 out of 4,918,783 ( 1.4%) loci
0.8: 110,040 out of 4,918,783 ( 2.2%) loci
0.9: 289,968 out of 4,918,783 ( 5.9%) loci
1.0: 4,180,722 out of 4,918,783 ( 85.0%) loci
IGV.js display of TR catalogs
Prior studies
[Willems 2014], one of the first studies to perform a genome-wide TR analysis using Illumina short read sequencing data, put considerable effort into defining an appropriate catalog, as described in their Results section and Supplementary Materials which are copy-pasted below. However, their approach was limited to the TR alleles observed within the hg19 reference genome. Also, they used simulations to calculate thresholds that distinguish biologically meaningful STRs from random or accidental repetative sequences, but this insilico approach is relatively blunt - just because a sequence could occur randomly with some probability doesn't mean it's not biologically important if it occurs invivo.
Also, data has since become available from the Human Pangenome Research Consortium (HPRC) as well as from other studies such as T2T-YAO from [He 2023] and the Illumina catalog by Qiu, Dolzhenko, et al. that now allows us to directly / empirically identify polymorphic TR loci and study their properties.
Relevant sections from the Results and Supplementary Materials of
Willems T, Gymrek M, Highnam G; 1000 Genomes Project Consortium, Mittelman D, Erlich Y. The landscape of human STR variation. Genome Res. 2014:
